Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
USP Standards for Labs: Compliance and Quality Guide
Unlock compliance with our guide, explaining USP standards for labs in the UK and EU. Enhance quality and stay ahead in testing and validation!
TL;DR:
- Many UK and European laboratories mistakenly believe USP standards only apply to American regulatory contexts, but these standards support EMA compliance and align with the European Pharmacopoeia, making them crucial for international research. Understanding when to validate, verify, or ensure system suitability—based on USP chapters 1225, 1226, and 1227—is essential for compliance and resource efficiency, especially during audits. Integrating USP standards into daily workflows and maintaining thorough documentation enhances global market access and demonstrates a high quality culture.
Many laboratory professionals in the UK and Europe assume USP (United States Pharmacopeia) standards are purely an American regulatory concern, applicable only to manufacturers supplying the FDA-regulated market. This assumption is both common and costly. In reality, USP methods support EMA compliance and harmonize with the European Pharmacopoeia (Ph.Eur.), making them directly relevant to any lab producing or testing research-grade products for international contexts. This guide clarifies the core USP standards, explains the critical distinction between method validation and verification, and outlines practical steps for sustained compliance in UK and EU laboratory settings.
Table of Contents
- What are USP standards and why do they matter for UK/EU labs?
- Core USP standards: Validation, verification, and system suitability
- When to validate, verify, or adapt: Navigating common scenarios
- Implementing and documenting USP compliance in everyday lab practice
- USP, EP, and international harmonization: The compliance advantage
- Why most labs misunderstand verification — and how to get it right
- Streamline your compliance journey with trusted lab solutions
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| USP standards boost compliance | Applying USP ensures international credibility and research-grade quality in UK/EU labs. |
| Verification streamlines lab work | Most labs can rely on method verification instead of full validation to save time and resources. |
| Document everything | Strong SOPs, records, and continual monitoring let labs pass audits and maintain standards. |
| Harmonization eases global access | Using both USP and EP helps labs meet FDA and EMA requirements for international product acceptance. |
What are USP standards and why do they matter for UK/EU labs?
The United States Pharmacopeia sets analytical and procedural benchmarks that define how laboratory methods must perform in terms of accuracy, precision, and reliability. While the FDA enforces these standards within the US, their influence extends far beyond American borders. For any lab engaged in producing, testing, or supplying research-grade materials internationally, alignment with USP is not optional. It is a practical foundation for global market access.
For UK and EU laboratories, the connection between USP and the European Pharmacopoeia is a key operational consideration. The two pharmacopeias have undergone substantial harmonization through the International Council for Harmonisation (ICH), meaning that many analytical procedures and acceptance criteria are now closely aligned. A lab that masters USP method requirements is simultaneously building competence in EP compliance, an advantage that pays dividends during audits with either EMA or MHRA.
The following are the primary reasons UK and EU labs should treat USP standards as core to their quality frameworks:
- Regulatory alignment: USP methods support EMA compliance and are harmonized with EP/Ph.Eur., covering major test categories including identity, purity, and potency.
- International research partnerships: Contract research organizations and academic collaborators working across US and EU jurisdictions require shared analytical frameworks to interpret results consistently.
- Audit readiness: Inspectors from major regulatory bodies recognize USP-aligned documentation as evidence of systematic, traceable quality management.
- Research-grade product credibility: For products such as sterile diluents, reconstitution solutions, and analytical reagents, adherence to recognized laboratory reagent standards supports claims of purity and consistency.
“USP standards may be legally enforceable in the US, but their practical authority in UK and EU laboratories derives from their role as the analytical lingua franca of global pharmaceutical and research-grade product quality.”
This credibility benefit is particularly significant for labs supplying materials to research institutions, universities, or commercial partners that operate across multiple regulatory jurisdictions simultaneously.
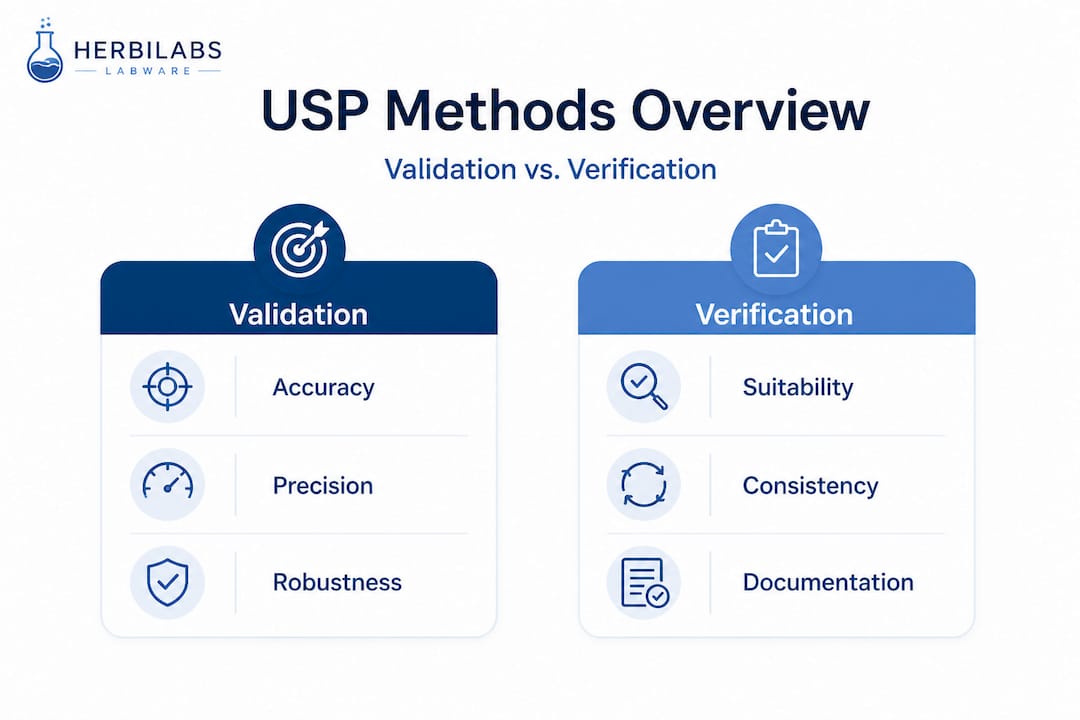
Core USP standards: Validation, verification, and system suitability
Understanding the distinction between the three core USP compendial chapters governing method performance is fundamental to correct implementation. The three chapters are: USP <1225> (Validation of Compendial Procedures), USP <1226> (Verification of Compendial Procedures), and USP <1227> (Validation of Alternative Procedures). Each serves a distinct function, and applying the wrong one to a given scenario is a frequent source of both over-expenditure and compliance risk.
USP <1225>: Method validation
USP <1225> defines validation parameters including accuracy (closeness to the true value), precision (encompassing repeatability, intermediate precision, and reproducibility), specificity (the ability to distinguish the analyte from other components), linearity and range, detection and quantitation limits, and robustness (tolerance to small deliberate variations in method conditions). Full validation under <1225> applies when a lab develops a new non-compendial method or introduces a significant modification to an existing compendial procedure.
USP <1226>: Method verification
For USP compendial methods used as-is, labs perform verification rather than full validation, confirming the method performs reliably under the lab’s specific equipment, personnel, and environmental conditions. Verification assesses a selected subset of validation parameters, typically accuracy and precision, making it a more focused and resource-efficient process than full validation.
USP <1227>: System suitability
<1227> addresses system suitability testing to ensure that analytical conditions are adequate immediately before and during each testing run. System suitability criteria must be met before results are accepted as valid, serving as a real-time checkpoint rather than a one-time establishment activity.

| Chapter | Purpose | When applied | Key parameters |
|---|---|---|---|
| <1225> | Full method validation | New or modified methods | Accuracy, precision, specificity, linearity, robustness |
| <1226> | Method verification | Compendial methods used as-is | Accuracy, precision (subset) |
| <1227> | System suitability | Before/during every analytical run | Resolution, tailing factor, RSD of replicate injections |
The following steps summarize a sound approach to selecting the appropriate chapter:
- Determine whether the method is pharmacopeial (compendial) or non-compendial.
- If compendial and used without modification, apply <1226> for verification.
- If compendial but modified, or if the method is non-compendial, proceed with full <1225> validation.
- Regardless of which chapter applies, establish and document system suitability criteria per <1227> for every routine run.
- Consult quality testing in biotech labs practices to align internal procedures with these requirements.
Pro Tip: When selecting parameters for verification under <1226>, assess the risk profile of your specific analyte and matrix. A high-risk impurity assay warrants testing more parameters than a general identity test, even though both technically fall under verification.
Achieving precision in quality control is not simply a function of instrument performance. It depends on how thoroughly each chapter’s requirements are embedded in day-to-day practice.
When to validate, verify, or adapt: Navigating common scenarios
With the three core chapters defined, the next challenge is mapping them correctly to real-world laboratory scenarios. This is where many QA teams encounter difficulty, because the boundaries between chapters are not always immediately obvious.
The primary decision point is whether a compendial method is being used exactly as written. If a lab adopts a USP or EP monograph method without any procedural modification, column substitution, solvent adjustment, or change in sample preparation, verification under <1226> is appropriate and sufficient. USP prioritizes verification over full validation for compendial methods precisely to streamline lab implementation while maintaining reliability under local conditions.
However, several scenarios trigger the requirement for full <1225> validation:
- Method modification: Changing any parameter of a compendial method, such as mobile phase composition or gradient profile, constitutes a modification requiring validation.
- Non-compendial methods: Methods developed in-house or sourced from scientific literature that have no pharmacopeial equivalent must be fully validated.
- Impurity and degradation product analysis: Tighter validation criteria for impurities apply compared to general assays, because the consequences of false negative impurity results can be severe. Detection and quantitation limits must be rigorously established.
- Non-standard matrices: When applying a compendial method to a matrix not covered by the original monograph, validation data must support the extension.
| Scenario | Required approach | Key documentation |
|---|---|---|
| Compendial method, no changes | Verification (<1226>) | Verification protocol, accuracy and precision data |
| Compendial method, modified | Validation (<1225>) | Full validation report, change control record |
| Non-compendial method | Validation (<1225>) | Development report, full validation, justification |
| Impurity analysis | Validation (<1225>) with tighter criteria | LOD, LOQ data, specificity data |
Pro Tip: During regulatory inspections, auditors frequently ask for the justification behind a lab’s choice of verification over validation. Prepare a brief written rationale for each method, cross-referencing the specific USP chapter criteria that support the decision. This document should sit alongside your SOP and be instantly retrievable.
Understanding these scenarios also supports smarter decisions about labware purity standards and reagent selection, since the quality of input materials directly affects whether method performance criteria can be met. Consistent operational efficiency in manufacturing contexts highlights the same principle: clarity in decision criteria eliminates rework.
Implementing and documenting USP compliance in everyday lab practice
Understanding USP requirements theoretically is one thing. Embedding them sustainably into laboratory operations is another. The following structured approach allows UK and EU labs to convert regulatory knowledge into practical, audit-ready workflows.
- Default to compendial methods: Use pharmacopeial procedures as your starting point. Labs must use compendial methods by default, validate or verify any adaptations, and establish SOPs that mirror USP structure for audit credibility.
- Develop USP-aligned SOPs: Each SOP should reference the applicable USP chapter, list acceptance criteria derived from validation or verification data, and specify system suitability requirements for each run.
- Implement formal change control: Any modification to a validated or verified method, even a minor instrument substitution, must be routed through a documented change control process before implementation.
- Train staff systematically: Training should cover not only the procedural steps but the underlying rationale for each acceptance criterion. Staff who understand why a system suitability test exists are more likely to respond correctly when it fails.
- Monitor method performance continuously: Ongoing monitoring and lifecycle management of analytical methods exceed one-time verification. Regular review of trending performance data allows labs to identify method drift before it causes out-of-specification results.
- Maintain complete documentation: Acceptance criteria derived statistically from performance data, aligned to product quality attributes, must be supported by robust documentation including protocols, raw data, and statistical summaries for audits.
Key documentation types for sustained USP compliance include:
- Validation or verification reports with raw data appendices
- System suitability results logged for every analytical run
- Change control records for all method modifications
- Staff training logs referencing specific USP chapter content
- Trending charts showing method performance over time
“Compliance is not a static state achieved at method implementation. It is a continuous process of monitoring, documenting, and responding to performance data in a manner that keeps the method fit for its intended purpose throughout its operational lifecycle.”
Using a lab quality control checklist calibrated to USP requirements helps teams maintain this discipline across multiple methods without relying on individual memory. Combining this with quality control tips for labs specific to research environments ensures that safe, reproducible science remains the foundation of every test run. Referencing equipment safety compliance steps alongside method documentation creates a complete operational record.
USP, EP, and international harmonization: The compliance advantage
For UK and EU labs with international research ambitions, the strategic value of adopting USP standards extends well beyond day-to-day compliance. It positions the laboratory as a credible, globally oriented facility capable of satisfying both FDA and EMA regulatory expectations without duplicating analytical effort.
The harmonization between USP and EP is most evident in major test categories such as dissolution, sterility, and particulate matter. Where ICH harmonization has been completed, a single validated method and dataset can often satisfy both pharmacopeial requirements simultaneously. This creates a direct efficiency gain for labs engaged in cross-border research or supplying materials to international partners.
The compliance advantages of dual USP/EP alignment include:
- Streamlined regulatory submissions: A common analytical package referencing both USP and EP chapters reduces the documentation burden for submissions to multiple agencies.
- Broader partner confidence: Research institutions and commercial collaborators operating under different jurisdictions can rely on shared analytical standards when evaluating data from a dual-compliant lab.
- Reduced audit friction: Inspectors from both EMA and MHRA recognize USP-aligned documentation, particularly for impurity profiling and method validation, as evidence of rigorous quality systems.
- Market access for research-grade products: USP alongside EP supports international research-grade compliance, bridging FDA and EMA expectations for labs supplying global markets.
“A UK laboratory that demonstrates competence in both USP and EP frameworks is not simply meeting two sets of requirements. It is communicating a quality culture capable of operating at the highest international standards.”
For labs working with reconstitution solutions, sterile diluents, or other research reagents, alignment with both frameworks is especially critical. Proper aseptic reagent preparation practices must be supported by validated methods that satisfy the most demanding compendial benchmarks regardless of the jurisdiction under which results will be reviewed.
Why most labs misunderstand verification — and how to get it right
The most persistent and resource-draining misconception we observe across quality-focused laboratories is the reflexive default to full method validation when verification is not only sufficient but explicitly preferred by USP. This tendency stems from a well-intentioned but misguided application of the precautionary principle. QA teams, concerned about audit risk, assume that more rigorous always means more defensible.
The reality is that USP designed <1226> precisely because full validation is disproportionate for compendial methods that have already undergone extensive development and validation by the pharmacopeia itself. When a lab runs a USP monograph method on its specific HPLC system with its own chromatographic column and trained analysts, the question is not whether the method is scientifically sound. It is whether the method performs reliably in that particular laboratory environment. Verification answers that question efficiently.
The practical cost of defaulting to full validation where verification applies is substantial. Validation studies for a single analytical method can consume weeks of analyst time and significant reagent resources. Multiplied across a lab’s full method portfolio, this misallocation of effort directly reduces the capacity available for actual research and testing.
The labs that get this right operate with a documented, risk-based decision framework that clearly maps each method to the appropriate USP chapter. They maintain concise written justifications for every verification decision, making their logic transparent to auditors. They recognize that using validated reagent standards for reliable results and well-characterized reference materials reduces verification risk without requiring the analytical burden of full validation.
The uncomfortable truth is that some labs perform full validation not because the science demands it, but because their QA leadership lacks confidence in the verification framework. Building that confidence through rigorous training and clear internal policy is a more effective investment than conducting unnecessary validation studies.
Streamline your compliance journey with trusted lab solutions
Navigating USP standards becomes significantly more manageable when your analytical inputs meet the purity and consistency standards these frameworks demand from the outset.
Herbilabs provides research-grade reagents, reconstitution solutions, and sterile diluents manufactured under strict quality controls that align with the benchmarks USP and EP compliance requires. Whether you are selecting the right materials when selecting lab reagents for peptide research, building confidence in your high-purity reagents, or evaluating options through a structured lab consumables comparison, Herbilabs offers the product quality and supporting resources that research-grade compliance demands. Our team supports UK and European labs with vetted materials, transparent specifications, and wholesale pricing for professional and institutional buyers.
Frequently asked questions
Do USP standards apply to research labs outside the US?
Yes, labs in the UK and Europe use USP standards to support compliance and harmonize with international requirements, especially for research-grade products, since USP methods support EMA compliance and are aligned with EP/Ph.Eur.
What is method verification under USP standards?
Method verification under USP <1226> confirms that a compendial procedure produces reliable results with a specific laboratory’s equipment, personnel, and conditions by testing a targeted subset of parameters such as accuracy and precision.
When is full validation required instead of verification?
Full validation is required when a lab modifies a compendial method, uses a non-compendial procedure, or conducts impurity analysis, because tighter criteria for impurities apply compared to general assays and non-standard methods must be fully characterized.
What documentation is needed for USP compliance?
Labs must maintain SOPs, change control logs, staff training records, raw analytical data, and statistical performance summaries, because robust documentation with protocols and raw data is required for regulatory audits.
How do USP and EP standards work together?
USP and EP are substantially harmonized through ICH, allowing labs to satisfy both FDA and EMA requirements with aligned analytical methods, since USP methods harmonize with EP/Ph.Eur. across major test categories for international research products.
Recommended
- Build the ultimate lab quality control checklist for 2026
- Best quality control tips for labs: ensure safe results
- Why rigorous quality testing matters in biotech labs
- Reagent quality control guide: ensure reliable lab results
- 21 CFR Part 11 Cybersecurity Compliance Guide | Symmetry Network Management