Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
Why rigorous quality testing matters in biotech labs
Learn why rigorous quality testing in biotech and pharma labs prevents recalls, contamination, and data failure. Practical QC steps for lab managers and researchers.
TL;DR:
- Rigorous quality testing ensures safety, efficacy, and purity of biotech and pharmaceutical products.
- Failures in QC, like data falsification or contamination, can lead to recalls and organizational collapse.
- Implementing structured SOPs, supplier validation, and trend analysis builds reliable laboratory quality systems.
When Able Laboratories falsified quality control records, the consequences reached far beyond regulatory fines: 3,184 batches were recalled, the company filed for bankruptcy, and 500 employees lost their jobs overnight. That single, systematic failure in testing rigor dismantled an entire organization. For independent researchers and laboratory managers working with bacteriostatic water, reagents, and reconstitution solutions, this is not a cautionary tale from a distant industry. It is a precise illustration of what happens when quality testing is treated as a formality rather than a scientific discipline. This guide covers the fundamentals of rigorous QC, the standards that govern it, the disasters that occur when it fails, and the practical steps labs can take to build lasting reliability.
Table of Contents
- What rigorous quality testing means in biotech and pharma
- Standards and protocols: USP, FDA, and microbial considerations
- What happens when rigorous QC fails: Lessons from disasters and recalls
- Building resilient QC: Practical steps for lab managers and researchers
- Perspective: Why most labs get quality testing wrong and how to do better
- Upgrade your quality testing with reliable reagents and labware
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| QC protects safety | Rigorous quality testing is the foundation for safe, effective biotech and pharmaceutical products. |
| Standards drive reliability | Adhering to USP, FDA, and microbial testing protocols prevents contamination and recalls. |
| Learn from failures | Analyzing real QC disasters helps labs avoid costly mistakes and strengthen procedures. |
| Implement best practices | Use actionable steps like SOPs, staff training, and supplier validation for ongoing QC success. |
What rigorous quality testing means in biotech and pharma
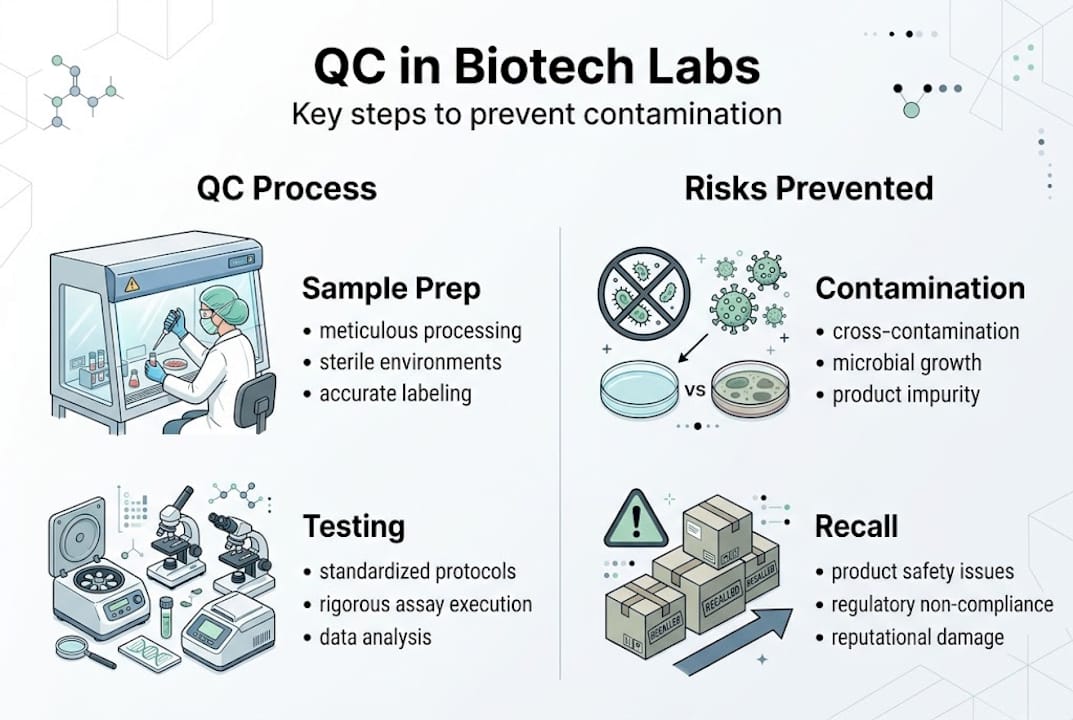
Quality testing in the biotech and pharmaceutical context refers to the structured, evidence-based process of verifying that every raw material, intermediate product, and finished batch meets defined specifications for safety, purity, identity, potency, and sterility. For bacteriostatic water and research reagents specifically, this means confirming that each lot is free of microbial contamination, endotoxins, and chemical impurities before it ever enters a reconstitution or assay workflow.
Rigorous quality testing in biotech and pharma ensures product safety, efficacy, purity, identity, and sterility, preventing patient harm from contaminated or substandard batches. That definition carries real operational weight. A reconstitution solution that passes a cursory visual inspection but harbors gram-negative bacterial endotoxins can invalidate an entire experimental series, trigger immune responses in in vivo models, or produce false assay signals that propagate through months of downstream data.
The core goals of a rigorous QC framework include:
- Safety: Confirming the absence of pathogens, toxic impurities, and pyrogenic contamination
- Efficacy: Verifying that reagents and diluents perform within validated ranges
- Sterility: Ensuring aseptic integrity throughout manufacturing and storage
- Purity: Quantifying and controlling contaminant levels below regulatory thresholds
- Identity: Confirming that each product is what its label claims it to be
The relationship between testing rigor and quality control fundamentals is direct: labs that implement structured QC programs reduce defect rates and waste substantially compared to those relying on informal inspection routines. Mature QC programs reduce defects by 50% and waste by 75%, according to FDA benchmarking data, figures that translate directly into cost savings and research reproducibility.
Pro Tip: Do not treat incoming material inspection as a single-point check. Build QC into every stage of reagent handling, from receipt through reconstitution, to catch deviation patterns before they compound.
For laboratory managers sourcing bacteriostatic water, the rigor of your supplier’s QC program is functionally inseparable from the rigor of your own. A supplier that cannot provide traceable test records, validated sterility data, and lot-specific certificates of analysis is introducing uncontrolled variability at the foundation of your research.

Standards and protocols: USP, FDA, and microbial considerations
Regulatory standards provide the technical baseline against which all quality testing is measured. In the United States and across most regulated research environments, the United States Pharmacopeia (USP) and the Food and Drug Administration (FDA) define the minimum acceptable testing requirements for biological reagents, sterile diluents, and aqueous solutions used in research and manufacturing.
Raw material testing, including for bacteriostatic water and reagents, is critical to prevent contamination entering production. The primary USP chapters governing this testing are:
| USP chapter | Test type | Primary objective |
|---|---|---|
| USP <61> | Microbial limits (non-sterile) | Quantify total aerobic microbial count |
| USP <62> | Specified microorganisms | Detect specific pathogens (e.g., E. coli, Salmonella) |
| USP <71> | Sterility testing | Confirm absence of viable microorganisms |
| USP <85> | Bacterial endotoxins (LAL) | Quantify endotoxin levels using limulus amebocyte lysate |
For bacteriostatic water and research reagents, USP <71> and <85> are particularly non-negotiable. Endotoxin contamination is especially insidious because standard sterilization methods, including autoclaving and filtration, do not reliably inactivate endotoxins. A lot that passes sterility testing can still carry pyrogenic load sufficient to disrupt cell-based assays or invalidate in vivo research.
Certificates of Analysis (CoAs) and USP-aligned sterility and endotoxin tests are essential for bacteriostatic water and reagents, since poor handling risks data irreproducibility. A CoA should document specific lot-level test results, not simply state that testing was performed. Researchers should request and critically review CoAs before accepting any shipment.
Best practices for preventing contamination at the raw material stage include:
- Quarantine all incoming materials pending release testing
- Verify supplier CoAs against in-house acceptance criteria
- Maintain documented traceability from lot receipt through final use
- Follow quality control tips for storage temperature, humidity, and container integrity
Pro Tip: Cross-reference supplier CoA results against your own in-house endotoxin testing on a rotating lot basis. Discrepancies between supplier and in-house data are early indicators of systemic supplier QC drift.
For labs operating at the interface of research and regulated manufacturing, sterility in manufacturing requires not only procedural compliance but also environmental monitoring and personnel training to close the gap between protocol and practice.
What happens when rigorous QC fails: Lessons from disasters and recalls
The history of biotech and pharmaceutical manufacturing contains repeated, expensive demonstrations of what happens when quality testing is inadequate, falsified, or structurally bypassed. These are not edge cases. They represent predictable outcomes of systemic weakness.
The Able Laboratories case remains one of the most documented examples of QC fraud in pharmaceutical manufacturing. Systematic data falsification led to the recall of 3,184 batches, corporate bankruptcy, and 500 job losses, a scale of destruction that traces directly to the decision to treat test records as administrative paperwork rather than scientific evidence.
In the diagnostics space, manufacturing contamination caused a Class I recall of Mesa Biotech’s Accula COVID test, resulting in false negatives with direct patient safety implications. Reagent contamination in UP-NIH kits similarly produced 30% indeterminate results before triggering recall actions.
| Incident | Root cause | Outcome |
|---|---|---|
| Able Laboratories | Data falsification in QC records | 3,184 batch recalls, bankruptcy, 500 job losses |
| Mesa Biotech Accula | Manufacturing contamination | Class I recall, false COVID-19 negatives |
| UP-NIH reagent kits | Reagent contamination | 30% indeterminate results, recall |
“The cost of inadequate quality testing is never limited to the failed batch. It propagates through every result generated from that batch, every decision made on that data, and every patient or researcher affected downstream.”
For laboratory managers, QC and research reliability are structurally linked: a single compromised reagent lot can corrupt an entire study’s dataset. The lessons from these incidents are sequential and actionable:
- Treat data integrity as a testable, auditable system, not an assumption
- Validate every raw material before it enters the production or assay workflow
- Investigate contamination incidents for root cause, not just batch disposition
- Maintain independent verification of supplier quality claims
- Build escalation pathways that remove the organizational incentive to suppress adverse results
Building resilient QC: Practical steps for lab managers and researchers
Moving from cautionary history to operational improvement requires a structured approach. The following steps represent field-tested practices for establishing QC programs that hold under the pressure of real laboratory conditions.
- Define and document SOPs: Every critical procedure, from reagent receipt through reconstitution and storage, must have a written, version-controlled Standard Operating Procedure (SOP). Undocumented practices are invisible to QC oversight.
- Validate supplier qualifications: Supplier audits, CoA review, and periodic in-house verification testing are not optional for regulated research environments. A lab QC checklist that includes supplier qualification criteria is a practical starting point.
- Train staff on data integrity principles: The FDA issued more than 160 warning letters between 2017 and 2022 specifically citing data integrity failures. Training is the upstream intervention.
- Monitor out-of-specification (OOS) rates: Routine testing typically yields 1 to 3% OOS results. Rates that trend above this range signal systemic issues requiring investigation, not just batch rejection.
- Implement trend analysis for recurring contamination: Sporadic contamination events are manageable. Recurring patterns of gram-negative contamination or particulate failures indicate process or environmental root causes that demand structural correction.
For bacteriostatic water and reagent handling specifically:
- Use only aseptic preparation techniques validated for your specific workflow
- Confirm container closure integrity before each use
- Log every reconstitution event with lot number, date, and preparer identity
- Source from suppliers who can provide certified lab supplies with traceable manufacturing documentation
Pro Tip: When contamination events occur, resist the pressure to close investigations with a single-cause explanation. Map the full contamination pathway: environment, personnel, materials, and equipment. Persistent patterns almost always have multiple contributing factors that single-cause investigations miss entirely.
Perspective: Why most labs get quality testing wrong and how to do better
The most common failure mode in laboratory QC is not ignorance of the standards. It is the structural tendency to treat compliance as the destination rather than the floor. Labs that invest heavily in finished-product testing while neglecting upstream controls, supplier qualification, and process trending are building a quality system that detects failures after they have already occurred, not one that prevents them.
The Rechon Life Science warning letter illustrates this precisely: persistent gram-negative and spore contamination required trend analysis and systemic process understanding, not surface-level investigations. The lab had investigation procedures. What it lacked was the analytical discipline to recognize and act on a recurring pattern.
The laboratories that consistently produce reproducible, reliable data are the ones that understand their processes deeply enough to anticipate where variability enters the system. They do not wait for OOS results to trigger action. They monitor trend lines, track environmental data, and question their reagent QC guide assumptions regularly. We would challenge every laboratory manager reading this to examine their own QC blind spots: Where is your system detecting failures rather than preventing them?
Upgrade your quality testing with reliable reagents and labware
Implementing the QC practices outlined here depends directly on the quality of the materials entering your workflow. If your bacteriostatic water, sterile diluents, or research reagents are not manufactured to documented purity standards with lot-specific testing, the rigor of your internal QC program is limited by that upstream variability.
Herbilabs supplies high-purity reagents and bacteriostatic water manufactured under strict quality controls, with full CoA documentation available for every lot. For laboratory managers evaluating sourcing options, our lab consumables comparison provides a structured framework for assessing supplier quality claims. Use our essential labware checklist to verify that your current toolkit meets the demands of reproducible, high-integrity research.
Frequently asked questions
What are the main risks of inadequate quality testing in biotech labs?
Inadequate testing exposes labs to contamination, unreliable experimental results, regulatory violations, product recalls, and direct patient harm. Quality testing failures in biotech and pharma have repeatedly demonstrated that substandard batches bypass detection and reach end users with serious consequences.
Why are USP <61>, <62>, <71>, and <85> important for reagent testing?
These chapters establish the validated methods for microbial limits, pathogen detection, sterility confirmation, and endotoxin quantification, covering the four primary contamination risks relevant to research reagents and bacteriostatic water. USP methods for microbial limits, sterility, and endotoxins form the non-negotiable baseline for compliant reagent testing.
How do quality failures impact a pharmaceutical company?
Quality failures trigger cascading consequences that extend well beyond the failed lot, including large-scale recalls, regulatory enforcement actions, reputational damage, and financial collapse. The Able Laboratories case, in which data falsification produced 3,184 batch recalls and bankruptcy, is the definitive reference point.
What practical steps improve lab quality control?
Establish version-controlled SOPs, qualify and audit suppliers, conduct regular data integrity training, monitor OOS trends, and perform root cause analysis on all contamination events. Mature QC programs that incorporate these elements consistently reduce defects by 50% and waste by 75% relative to less structured approaches.