Private Label, White Label, Wholesale partnerships available - EU, USA and UK - Free shipping from €75
Sample Preparation Tutorial for Lab Professionals 2026
Elevate your lab skills with our sample preparation tutorial! Learn essential workflows and techniques for accurate analytical results in 2026.
TL;DR:
- Sample preparation transforms complex samples into clean, reproducible forms crucial for accurate analysis; errors in this step can propagate bias and distort results. Proper execution of steps like sampling, homogenization, extraction, cleanup, and concentration ensures data integrity, with instrument-specific protocols and quality controls being essential. Consistent training, contamination prevention, and validated materials are vital to maintaining reliable, high-quality analytical results.
Sample preparation is defined as the systematic process of transforming complex, raw matrices into clean, instrument-compatible forms suitable for precise and reproducible analytical measurement. Every downstream result depends on the quality of this step. Preparation errors propagate as systematic bias, corrupting calibration curves and distorting quantitative outputs across an entire analytical batch. Whether you work with ICP-MS, LC-MS, or HPLC, the integrity of your data is determined before the instrument ever fires. This sample preparation tutorial covers the complete workflow, instrument-specific optimization, common failure modes, and the materials that support reliable results.
What are the essential steps in a sample preparation workflow?
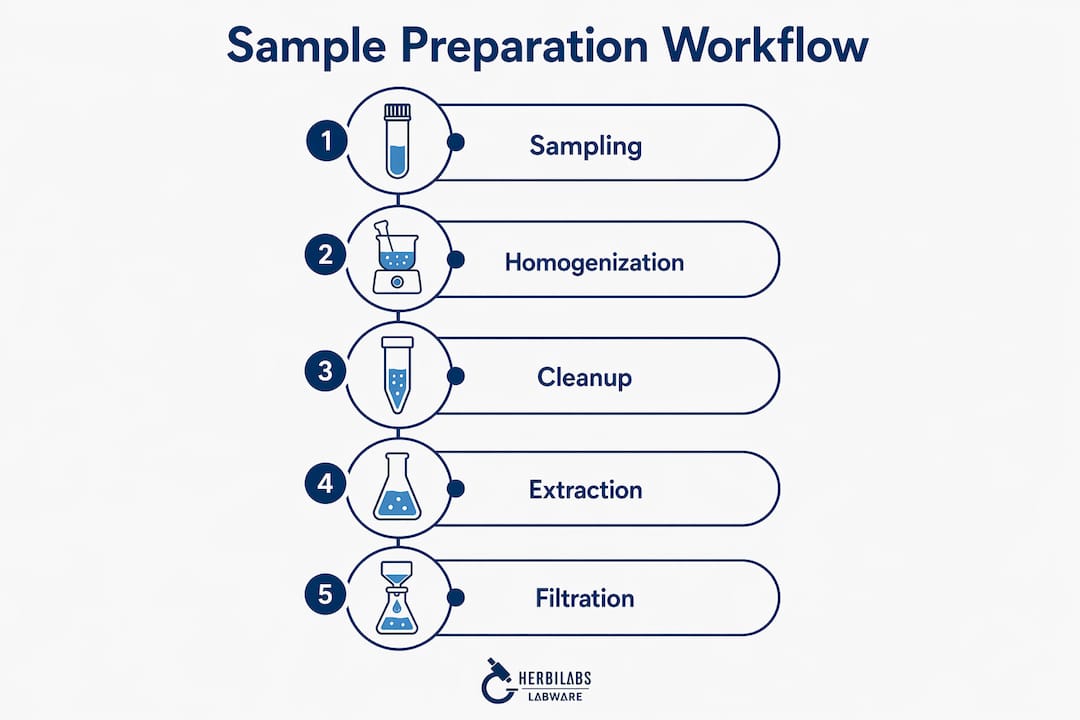
Representative sampling, homogenization, extraction, cleanup, and concentration form the standard sequence for converting a complex matrix into an analyte-ready solution. Each step has a defined purpose, and skipping or shortcutting any one of them introduces error that cannot be corrected later. The following numbered sequence applies to most analytical contexts, though the specific techniques at each stage vary by matrix and instrument.
- Representative sampling. Select aliquots that are statistically valid for the bulk material. Inhomogeneous sampling is the most common source of pre-analytical error in environmental and biological matrices.
- Homogenization. Mechanical or chemical homogenization achieves uniform analyte distribution. Bead milling, probe sonication, and rotor-stator homogenizers are standard tools depending on sample viscosity and fragility.
- Extraction. Transfer the target analyte from the matrix into a workable solvent or phase. Liquid-liquid extraction (LLE), solid-phase extraction (SPE), and Soxhlet extraction each suit different analyte polarities and matrix types.
- Cleanup and purification. Remove co-extracted interferences, particulates, and matrix components that would suppress signals or damage instrumentation. SPE cartridges, protein precipitation, and spin filters are the primary tools at this stage.
- Concentration or dilution. Adjust analyte concentration to fall within the instrument’s linear dynamic range. Nitrogen evaporation, rotary evaporation, and volumetric dilution with calibrated pipettes are the most common approaches.
- Derivatization (where required). Convert analytes to forms compatible with detection, such as silylation for GC-MS or fluorescent tagging for fluorescence detection in HPLC.
| Step | Primary action | Goal |
|---|---|---|
| Representative sampling | Collect statistically valid aliquots | Eliminate sampling bias |
| Homogenization | Mechanically or chemically blend sample | Achieve uniform analyte distribution |
| Extraction | LLE, SPE, or Soxhlet transfer | Isolate target analyte from matrix |
| Cleanup | SPE, spin filtration, precipitation | Remove interferences and particulates |
| Concentration/dilution | Evaporation or volumetric dilution | Match instrument detection range |
| Derivatization | Chemical modification of analyte | Improve instrument compatibility |
Pro Tip: Balance method complexity against analyte recovery at every design stage. Each additional cleanup step introduces a potential loss point. Validate recovery at the lowest expected concentration before committing to a multi-step protocol.
How to select and optimize techniques for different instruments
Instrument type is the primary variable that determines which sample prep methods and acceptance criteria apply. A protocol optimized for ICP-MS will actively harm an LC-MS run if applied without modification.

ICP and ICP-MS preparation
For ICP and ICP-MS workflows, acid concentration must remain below 5% v/v in the final solution to avoid signal drift and plasma instability. This is a hard limit, not a guideline. When hydrofluoric acid (HF) is required for silicate digestion, quartz components in the sample introduction system must be replaced with inert alternatives such as PTFE or sapphire, because HF dissolves quartz and contaminates the analyte stream. Matrix composition directly controls background signal and detection limits in ICP-MS, making matrix-matched calibration standards mandatory for high-accuracy work.
LC-MS proteomics preparation
LC-MS proteomics workflows require aggressive cleanup because detergents, salts, and lipids suppress ionization and degrade spectra quality. Desalting with commercial traps at approximately 0.5 µL bed volume removes these interferences while minimizing sample loss. Detergent choice at the lysis stage is not trivial. SDS produces superior protein extraction but requires complete removal before LC-MS injection. Alternatives such as RapiGest SF or n-dodecyl-β-D-maltoside are MS-compatible and reduce the cleanup burden significantly.
HPLC preparation
HPLC mobile phase preparation demands volumetric accuracy and filtration through 0.22 µm membranes to remove particulates that would block frits and damage columns. System suitability tests must be performed before each analytical sequence, with acceptance criteria of %RSD ≤1% for triplicate injections of a reference standard. Suitability results are valid for a maximum of 24 hours, and prepared mobile phase should be discarded after 7 days or immediately upon any sign of haziness or microbial growth.
| Instrument | Key prep requirement | Critical acceptance criterion |
|---|---|---|
| ICP/ICP-MS | Acid concentration control, inert materials for HF | Residual acid below 5% v/v |
| LC-MS proteomics | Desalting, detergent removal | Clean baseline, no ion suppression |
| HPLC | Mobile phase filtration, system suitability | %RSD ≤1% for triplicate injections |
Pro Tip: For trace-level ICP-MS work, use only PTFE or PFA vials and ultrapure reagents at sub-ppb blank levels. Borosilicate glass leaches boron and sodium into acidic solutions, elevating background at masses 10, 11, and 23.
What common mistakes undermine sample prep reliability?
The most damaging errors in sample preparation are not dramatic failures. They are systematic, repeatable, and invisible until a reference material or inter-laboratory comparison exposes them. Understanding the failure modes is the first step toward prevention.
Proper preparation eliminates systematic bias and protects instruments from damage, but only when each stage is executed with discipline. The following errors account for the majority of reproducibility failures in analytical laboratories.
- Contamination from glassware. Inadequately cleaned volumetric flasks and pipettes introduce trace metals, organic residues, and detergent films. Acid-washing followed by ultrapure water rinsing is the minimum standard for trace analysis. Refer to a structured contamination prevention protocol to formalize this process.
- Impure reagents. Using analytical-grade solvents where LC-MS-grade or trace-metal-grade reagents are required introduces background interferences that mimic or suppress analyte signals.
- Overcomplicated workflows. Each additional extraction or cleanup step adds a recovery loss point. A method with six steps and 70% recovery at each step delivers less than 12% overall recovery, which is analytically unacceptable.
- Particulates in the final solution. Unfiltered samples damage HPLC frits, block ICP nebulizers, and cause irreproducible injection volumes. All final solutions should pass through a 0.22 µm syringe filter before injection.
- Inconsistent homogenization. Applying different sonication times or bead-milling speeds across a batch creates between-sample variability that cannot be distinguished from true biological or chemical differences.
- Improper spin filter technique. Spin filters require membrane pre-rinsing and centrifugation for 10 to 30 minutes, followed by inversion and re-spinning to concentrate larger analytes retained on the membrane. Skipping the pre-rinse step introduces glycerol and membrane-derived contaminants into the filtrate.
Pro Tip: Run a method blank through every preparation batch. A blank that shows signal indicates contamination at a specific step. Systematically removing steps from the blank protocol identifies the contamination source faster than any other diagnostic approach.
What tools and materials support optimal sample preparation?
The quality of sample preparation output is constrained by the quality of the materials and equipment used. High-purity reagents, calibrated labware, and properly maintained instrumentation are not optional upgrades. They are the baseline for defensible analytical data.
Selecting appropriate labware for your workflow affects both contamination risk and measurement accuracy. Class A volumetric flasks and calibrated micropipettes are the minimum standard for quantitative work. Centrifuges used for spin filtration must be validated for speed accuracy, as rotational variability directly affects membrane performance and filtrate clarity.
| Material or equipment | Function | Key specification |
|---|---|---|
| Class A volumetric flasks | Accurate dilution and standard preparation | Calibrated to ±0.05% tolerance |
| Calibrated micropipettes | Precise volume transfer | Verify with gravimetric check quarterly |
| Spin/syringe filters (0.22 µm) | Remove particulates from final solution | Membrane material matched to solvent |
| Centrifuge | Separation and spin filtration | Speed accuracy ±2% of set value |
| Ultrapure water system | Solvent and reagent preparation | Resistivity ≥18.2 MΩ·cm at 25°C |
| High-purity acids and solvents | Digestion, extraction, mobile phase | Trace-metal grade or LC-MS grade |
Beyond equipment, quality control practices define whether a preparation method is fit for purpose. Batch controls, including a method blank, a matrix-matched spike, and a certified reference material, must accompany every analytical sequence. System suitability and batch controls provide the documented evidence that the preparation method performed within validated parameters on the day of analysis.
- Use ultrapure water with resistivity ≥18.2 MΩ·cm for all reagent preparation and final dilutions. The role of high-purity water in reducing background signal is well established across ICP-MS and LC-MS platforms.
- Store reagents in their original containers, away from direct light and heat, and label with the date of opening and expiration.
- Dispose of mobile phase after 7 days regardless of appearance, and discard any reagent showing turbidity, precipitation, or off-color immediately.
Key takeaways
Accurate sample preparation requires a structured, instrument-specific workflow where every step from representative sampling through final filtration is executed with validated materials and documented quality controls.
| Point | Details |
|---|---|
| Systematic workflow is mandatory | Follow the sequence: sampling, homogenization, extraction, cleanup, and concentration without skipping steps. |
| Instrument type dictates method | ICP-MS, LC-MS, and HPLC each require distinct cleanup criteria and acceptance thresholds. |
| Contamination is the primary failure mode | Use trace-metal-grade reagents, acid-washed glassware, and 0.22 µm filtration for all final solutions. |
| Spin filter technique matters | Pre-rinse membranes, centrifuge 10 to 30 minutes, and re-spin inverted to recover retained analytes. |
| Quality controls validate every batch | Run method blanks, matrix spikes, and certified reference materials with every analytical sequence. |
Why sample prep deserves the same rigor as the analysis itself
Most reproducibility failures I encounter trace back to the same root cause: sample preparation was treated as a routine processing step rather than an integral part of the analytical method. The instrument gets calibrated, the software gets validated, and the column gets tested. The sample prep protocol gets assumed.
Sample preparation must be viewed as an analytical-method integrity step, not a precursor to the real work. This distinction changes how you allocate time, reagent budget, and validation effort. In my experience, the laboratories with the most reproducible data are not the ones with the most sophisticated instruments. They are the ones with the most disciplined preparation protocols, written down, followed consistently, and reviewed when results drift.
Simplicity is underrated. A three-step preparation method with 95% recovery and a %RSD of 0.8% outperforms a six-step method with 60% recovery and a %RSD of 3.5% every time. Complexity creates variability. Variability creates bias. Bias invalidates data. The goal is not the most thorough cleanup possible. The goal is the minimum cleanup necessary to achieve the required accuracy and precision, executed identically every time.
Preventive quality control is more effective than corrective troubleshooting. Running a method blank at the start of every batch takes five minutes. Diagnosing a contamination event after 40 samples have been processed takes days. The investment in prevention is not optional for laboratories that produce data others rely on.
— Ragnar
Explore Herbilabs reconstitution solutions for your sample prep workflow
Precise sample preparation depends on the quality of every reagent in the protocol, and reconstitution solutions are no exception. Herbilabs supplies research-grade reconstitution solutions and sterile diluents manufactured to strict purity standards, specifically designed to support demanding analytical workflows where contamination and inconsistency are not acceptable.
For researchers working with lyophilized peptides or sensitive biological samples, Herbilabs’ high-purity reconstitution solutions provide the clean, stable diluent matrix that accurate reconstitution requires. The product range is updated for 2026 and includes options across multiple volumes to match your protocol scale. For guidance on selecting the right reagents for your specific analytical context, the Herbilabs laboratory reagent selection guide provides a structured framework aligned with current best practices.
FAQ
What is sample preparation in analytical chemistry?
Sample preparation is the process of converting a raw, complex sample into a clean, instrument-compatible form for accurate analytical measurement. It includes steps such as homogenization, extraction, cleanup, and concentration or dilution.
Why does acid concentration matter for ICP-MS samples?
Residual acid concentration above 5% v/v causes plasma instability, signal drift, and elevated background in ICP-MS analysis. Controlling final acid concentration is one of the most critical parameters in ICP-MS sample prep method design.
How do spin filters work in sample preparation?
Spin filters use centrifugal force to pass small molecules through a size-selective membrane while retaining larger species. Pre-rinsing the membrane and centrifuging for 10 to 30 minutes, followed by an inverted re-spin, maximizes analyte recovery and minimizes membrane-derived contamination.
What is the %RSD acceptance criterion for HPLC system suitability?
HPLC system suitability requires a %RSD of ≤1% for triplicate injections of a reference standard. Results are valid for a maximum of 24 hours, after which the test must be repeated before continuing analysis.
How do you prevent contamination during sample preparation?
Use acid-washed glassware, trace-metal-grade or LC-MS-grade reagents, and 0.22 µm filtered final solutions. Running a method blank with every batch identifies contamination sources before they affect sample results.