TL;DR:
- Aseptic manufacturing prevents microbial contamination during production of thermolabile biologics.
- It relies on strict environmental control, validation, and barrier technologies to maintain sterility.
- Continuous process validation and evolving regulations demand a proactive approach to contamination control.
A single undetected microbial contaminant introduced during the filling of a lyophilized biologic can render an entire batch non-sterile, triggering costly recalls, regulatory action, and potential patient harm. In aseptic manufacturing, that risk is not theoretical; it is the central challenge that every process step, environmental control, and validation protocol is designed to prevent. Unlike terminal sterilization, aseptic processing demands that sterility be maintained continuously, not achieved at the end of a production cycle. This article covers the core principles, methodologies, validation requirements, and emerging technologies that define modern aseptic manufacturing, providing laboratory professionals and researchers with the technical depth needed to strengthen their own sterility assurance practices.
Table of Contents
- Defining aseptic manufacturing: Core principles and why it matters
- Key components and methodologies in aseptic manufacturing
- Validation and contamination control: Ensuring ongoing sterility
- Aseptic vs terminal sterilization: Choosing the right method
- Challenges and future trends: Automation, barriers, and regulatory expectations
- Expert perspective: Why aseptic sterility is a moving target in modern labs
- Advance your lab’s sterility: Proven resources and solutions
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Aseptic vs terminal sterilization | Aseptic manufacturing is essential for heat-sensitive drugs; terminal sterilization is safer but not always an option. |
| Validation is ongoing | Sterility validation through media fills and monitoring is a recurrent process, not a one-time event. |
| Barrier technologies reduce risk | Isolators, RABS, and automation are effective at minimizing contamination from human contact. |
| Lab culture matters | Sterility assurance requires commitment at every level and adaptation as technology and regulations evolve. |
Defining aseptic manufacturing: Core principles and why it matters
Aseptic manufacturing is, at its most precise, the process where sterile drug product, containers, and closures are sterilized separately and then brought together in a controlled sterile environment to prevent microbial contamination. This definition immediately distinguishes aseptic processing from terminal sterilization, where the finished, sealed product is sterilized as a whole unit after assembly. The distinction matters enormously in practice because many modern biologics, including monoclonal antibodies, peptide-based therapeutics, and recombinant proteins, are thermolabile, meaning they degrade irreversibly when exposed to the heat or radiation required for terminal sterilization.
The primary purpose of aseptic manufacturing is therefore to enable the production of sterile dosage forms that cannot survive conventional sterilization cycles. This includes not only injectable biologics but also ophthalmic solutions, certain small-molecule injectables, and reconstitution diluents used in research and clinical settings. The stakes extend well beyond product efficacy. Microbial contamination in a sterile injectable can cause septicemia, endotoxin-mediated pyrogenic reactions, or localized infections, outcomes that are directly attributable to failures in aseptic process control.
Key reasons aseptic manufacturing is required across biotech and pharmaceutical sectors include:
- Thermolabile products: Many biologics and peptides cannot withstand autoclaving or dry heat sterilization without structural degradation.
- Regulatory mandate: FDA 21 CFR Part 211 and EU GMP Annex 1 require aseptic processing for products that cannot be terminally sterilized.
- Product integrity: Aseptic conditions preserve the chemical and biological activity of sensitive active pharmaceutical ingredients (APIs).
- Patient safety: Injectable products administered without a terminal kill step carry zero tolerance for microbial contamination.
- Reconstitution solutions: Diluents such as bacteriostatic water for injection must be manufactured aseptically to maintain their sterility throughout shelf life.
“Aseptic processing is not simply a cleaner version of standard manufacturing. It is a fundamentally different paradigm where every material, surface, and action within the production zone must be treated as a potential contamination vector.”
For laboratory professionals who work with reconstitution reagents or conduct peptide research, understanding the aseptic techniques checklist that governs these processes is directly relevant to maintaining the integrity of experimental outcomes. Regulatory oversight from both the FDA and the European Medicines Agency (EMA) continues to tighten expectations around contamination control strategy (CCS), making foundational knowledge of aseptic principles increasingly essential.
Key components and methodologies in aseptic manufacturing
The operational backbone of aseptic manufacturing rests on a set of interdependent components, each of which must function correctly for the overall system to maintain sterility. Key methodologies include component sterilization, Grade A cleanroom environments with unidirectional HEPA airflow, aseptic assembly and filling, environmental and personnel monitoring, and media fills for validation. Failure in any single element propagates risk across the entire process chain.
The standard sequence of operations in an aseptic fill-finish line follows a defined order:
- Component preparation and sterilization: Vials, stoppers, and closures are depyrogenated and sterilized using dry heat tunnels or autoclaves before entering the cleanroom.
- Bulk product sterilization: The drug solution is sterile-filtered through 0.22-micron membrane filters to remove bioburden prior to filling.
- Environmental preparation: The Grade A filling zone is purged with unidirectional HEPA-filtered airflow, and surface bioburden is reduced through validated cleaning and disinfection cycles.
- Aseptic assembly and filling: Product is filled into sterilized containers under Grade A conditions, with operators or automated systems working within the critical zone.
- Environmental and personnel monitoring: Viable and non-viable particle counts are measured continuously during filling operations to detect excursions in real time.
- Media fill validation: Nutrient growth medium replaces the actual product to simulate the aseptic process and confirm that the system can consistently produce sterile units.
| Cleanroom grade | ISO equivalent | Maximum particles (0.5 µm/m³) | Typical application |
|---|---|---|---|
| Grade A | ISO 5 | 3,520 | Filling zone, critical operations |
| Grade B | ISO 5 (at rest) | 3,520 | Background to Grade A |
| Grade C | ISO 7 | 352,000 | Less critical aseptic preparation |
| Grade D | ISO 8 | 3,520,000 | Gowning, material preparation |
Barrier technologies play an increasingly central role in modern aseptic manufacturing. Restricted Access Barrier Systems (RABS) and isolators physically separate operators from the critical processing zone, reducing the frequency and impact of human interventions. Isolators, in particular, provide the highest level of separation by maintaining a closed, positively pressurized environment that is independently sterilized using vaporized hydrogen peroxide (VHP) before each campaign.

Pro Tip: When evaluating your facility’s Grade A zone, verify that unidirectional airflow velocity is maintained at 0.36 to 0.54 m/s at the work surface. Deviations outside this range can create turbulent zones that transport particles toward open containers.
For practical quality control tips applicable to smaller-scale laboratory settings, the same principles of environmental monitoring and controlled access apply, even when full pharmaceutical-grade infrastructure is not available.
Validation and contamination control: Ensuring ongoing sterility
Validation is the formal, documented evidence that an aseptic process consistently produces sterile product. The media fill, also called a process simulation test, is the cornerstone of this validation framework. In a media fill, a microbiological growth medium such as soybean casein digest (SCDB) replaces the actual drug product and is processed through the full aseptic filling sequence. Incubated units are then examined for turbidity, which indicates microbial growth.
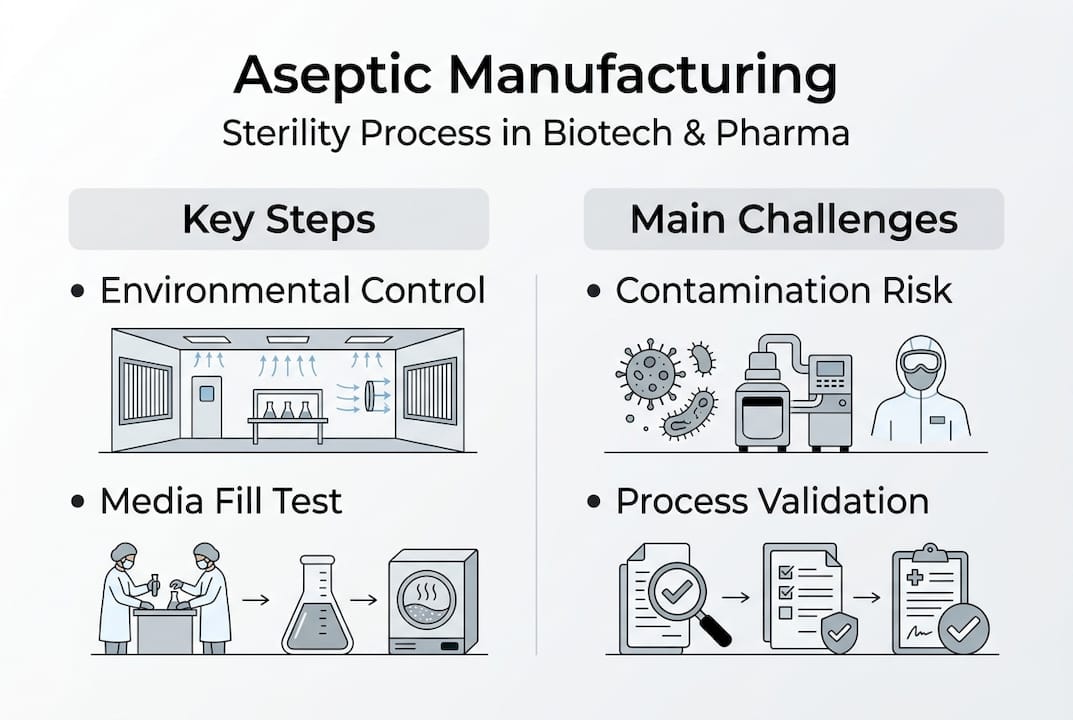
The FDA’s media fill benchmarks define acceptable contamination rates by batch size: for runs below 5,000 units, zero contaminated units are acceptable; one contaminated unit triggers investigation and repeat testing, while two require revalidation. For runs between 5,000 and 10,000 units, one contaminated unit requires revalidation, and two or more require revalidation with root cause analysis. For runs exceeding 10,000 units, a contamination rate of no more than 0.1% is targeted.
Personnel represent the single greatest contamination risk in any aseptic operation. Rigorous training, gowning qualification, and minimized interventions are critical controls because human skin, breath, and movement continuously generate viable particles. Gowning qualification requires operators to demonstrate that their gowning technique does not itself introduce contamination, typically assessed through glove and suit surface sampling after gowning.
Core contamination control practices include:
- Continuous environmental monitoring (EM): Settle plates, active air samplers, and contact plates are deployed throughout filling operations to provide real-time bioburden data.
- Intervention classification: All manual interventions during filling are classified as inherent (planned) or corrective (unplanned), with corrective interventions triggering immediate review.
- Disinfectant rotation: Using two or more disinfectants with different mechanisms of action prevents the development of resistant microbial populations on cleanroom surfaces.
- Contamination Control Strategy (CCS): EU GMP Annex 1 (2022 revision) now mandates a holistic CCS document that maps all contamination risks and their corresponding controls across the entire manufacturing site.
Pro Tip: Incorporate worst-case interventions into every media fill run. Regulatory inspectors specifically evaluate whether simulated interventions reflect the most challenging real-world scenarios encountered during production.
Maintaining labware integrity steps and following validated procedures for handling lab reagents are directly analogous to these pharmaceutical-grade controls, particularly when working with sterile reconstitution solutions in research environments.
Aseptic vs terminal sterilization: Choosing the right method
The choice between aseptic processing and terminal sterilization is not simply a matter of preference; it is a risk-based decision driven by product characteristics, regulatory expectations, and manufacturing capability. Terminal sterilization is always preferred when feasible because it achieves a Sterility Assurance Level (SAL) of 10^-6, meaning the probability of a non-sterile unit surviving the process is one in one million. Aseptic processing does not achieve a defined SAL in the same sense because sterility relies on process controls rather than a terminal kill step.
| Attribute | Aseptic processing | Terminal sterilization |
|---|---|---|
| Sterility mechanism | Process control throughout | Post-assembly kill step |
| SAL achieved | Process-dependent | 10^-6 |
| Suitable product types | Thermolabile biologics, peptides | Heat-stable small molecules |
| Contamination risk | Higher (no final kill step) | Lower |
| Regulatory preference | When terminal not feasible | Preferred by FDA/EMA |
| Cost and complexity | Higher | Lower |
Aseptic processing carries inherently higher contamination risk because there is no post-assembly sterilization event to eliminate any contamination introduced during filling. This is why regulatory agencies including the FDA explicitly state that terminal sterilization should be used wherever the product can tolerate it. Aseptic processing is reserved for products where terminal sterilization is not feasible, including most monoclonal antibodies, lyophilized peptide formulations, and complex biologics.
For researchers working with bacteriostatic vs sterile water as reconstitution vehicles, understanding this distinction clarifies why the manufacturing method of the diluent itself matters. Bacteriostatic water for injection is produced aseptically because the benzyl alcohol preservative system and the water itself cannot undergo terminal sterilization without compromising the product’s chemical profile.
The decision framework is straightforward: if a product can withstand moist heat (121°C, 15 minutes), dry heat, radiation, or ethylene oxide sterilization without degradation, terminal sterilization is the appropriate choice. If not, aseptic processing is required, and the full weight of environmental controls, personnel training, and validation must be applied rigorously.
Challenges and future trends: Automation, barriers, and regulatory expectations
Aseptic manufacturing faces a set of persistent operational challenges that no amount of regulatory guidance fully resolves. Human interventions remain the primary contamination risk, and the use of isolators and RABS to minimize operator contact is now considered best practice. Beyond personnel, supply chain disruptions for critical components such as stoppers and filters, cold chain requirements for temperature-sensitive biologics, and the high capital cost of Grade A infrastructure represent ongoing constraints for both large manufacturers and smaller research facilities.
Key challenges currently facing aseptic manufacturing operations include:
- Cold chain integrity: Many biologics require continuous 2 to 8°C storage from bulk production through fill-finish, adding complexity to environmental control and logistics.
- Component supply variability: Shortages of sterilized stoppers, vials, and filter membranes can force process changes that require revalidation.
- Regulatory evolution: The 2022 revision of EU GMP Annex 1 introduced the mandatory CCS requirement and raised expectations for barrier technology adoption, requiring significant process updates across the industry.
- Cost of compliance: Maintaining Grade A environments, qualified personnel, and validated processes represents a substantial and ongoing financial commitment.
“The trajectory of aseptic manufacturing is clearly toward greater automation and physical separation of operators from critical zones. Robotics and closed systems are not optional upgrades; they are the direction that both regulators and industry leaders are moving.”
The trend toward robotics and closed systems to reduce human error and meet updated Annex 1 requirements is accelerating. Robotic filling systems, automated visual inspection, and closed-vial filling technologies are being adopted at scale, particularly in facilities producing high-value biologics where the cost of a contamination event far exceeds the capital investment in automation. For laboratory professionals, understanding these trends informs decisions about facility design, equipment procurement, and the SOPs needed to avoid contamination mistakes during reagent preparation and reconstitution.
Expert perspective: Why aseptic sterility is a moving target in modern labs
One of the most persistent misconceptions in aseptic manufacturing is that a successful validation event, whether a passing media fill or a clean regulatory inspection, represents a stable, achieved state. It does not. Sterility is a dynamic property of a system, not a fixed characteristic of a product. Personnel change, equipment ages, environmental conditions shift seasonally, and new materials introduce uncharacterized bioburden. Each of these variables can erode a previously validated state without triggering an immediate, detectable signal.
The laboratories and manufacturers that maintain the strongest sterility records are those that treat their contamination control strategy as a living document, revised in response to trending data, near-miss events, and regulatory feedback, rather than a compliance artifact filed after initial validation. Adapting SOPs based on environmental monitoring trends, rather than waiting for an excursion, reflects the mindset shift from compliance to genuine quality culture.
For researchers relying on aseptically manufactured reagents, this perspective is directly relevant. The quality of a reconstitution diluent is only as reliable as the ongoing rigor of the manufacturing process that produced it. Reviewing modern aseptic practices and applying them consistently in your own laboratory environment is not optional; it is the foundation of reproducible, trustworthy research.
Advance your lab’s sterility: Proven resources and solutions
Strengthening aseptic practices in your laboratory requires both the right knowledge framework and access to reagents manufactured to verifiable sterility standards. Herbilabs provides research-grade bacteriostatic water and sterile diluents produced under strict aseptic manufacturing conditions, supported by rigorous quality control protocols designed for demanding research environments.
Explore our aseptic preparation guide for step-by-step protocols applicable to reagent handling and peptide reconstitution. When sourcing diluents, our resource on selecting lab reagents provides a structured framework for evaluating purity, compatibility, and manufacturing standards. For a deeper understanding of why manufacturing quality directly affects research outcomes, the guide on high-purity reagents connects manufacturing rigor to experimental reproducibility.
Frequently asked questions
What products require aseptic manufacturing instead of terminal sterilization?
Aseptic manufacturing is necessary for heat-sensitive biologics and pharmaceuticals that would be degraded or inactivated by the temperatures, radiation, or chemical agents used in terminal sterilization processes.
What is a media fill and why is it important?
A media fill is a process simulation test in which a microbiological growth medium replaces the drug product to validate that the aseptic filling process consistently produces sterile units under real operating conditions, including planned and unplanned interventions.
How do barrier technologies like isolators and RABS reduce contamination risk?
Isolators and RABS physically separate operators from the critical filling zone, directly reducing the frequency of human-introduced contamination, which FDA and EU GMP identify as the primary source of microbial risk in aseptic operations.
What is Sterility Assurance Level (SAL) and how does it differ between methods?
SAL is the probability of a single non-sterile unit remaining after a sterilization process; terminal sterilization achieves SAL 10^-6, while aseptic processing relies on validated process controls rather than a defined probabilistic kill step.
Recommended
- Aseptic Techniques During Reagent Preparation – A PRO Researcher’s 1# Checklist – Herbilabs Labware
- Best quality control tips for labs: ensure safe results
- Lab Best Practices 2025 for Handling and Storage of Bacteriostatic Water – Herbilabs Labware
- Bacteriostatic vs Sterile Water: Safe Lab Application Guide 2025 – Herbilabs Labware


